
Exufiber nella pratica
In uno studio su 21 pazienti trattati con Exufiber®, la condizione della cute perilesionale è migliorata e le dimensioni delle ulcere del piede diabetico si sono ridotte.
Studio post-clinico multicentrico, in aperto, non comparativo sulle prestazioni e la sicurezza di una medicazione in fibra gelificante per il trattamento delle ulcere del piede diabetico
Chadwick, P., McCardle, J. Journal of Wound Care 2016; 25(4):290-300.
- L’ulcerazione del piede nei pazienti diabetici è difficile da gestire.
- I pazienti con diabete scarsamente controllato sono a maggiore rischio di ulcerazione e amputazione del piede.
- La scelta della medicazione è un aspetto importante della gestione delle ulcere del piede diabetico (DFU).
- Le DFU sono associate a importanti costi di natura clinica, personale, sociale ed economica.
- In maniera analoga ad altre ferite croniche, le DFU possono produrre eccessivi livelli di essudato; di conseguenza, la cute perilesionale è a rischio di macerazione.
Obiettivo
Valutare le prestazioni e la sicurezza della medicazione in fibra gelificante Exufiber nel trattamento delle ulcere del piede diabetico.
Metodi
- Si tratta di uno studio aperto, non comparativo, multicentrico.
- Sono stati reputati idonei sia i pazienti in regime ambulatoriale sia i pazienti in regime di ricovero.
Obiettivo primario dello studio:
- Valutare le prestazioni e la sicurezza di Exufiber quando utilizzata secondo le indicazioni sulle ulcere del piede diabetico.
- In linea con tale obiettivo, sono stati misurati diversi parametri al fine di monitorare la condizione della cute perilesionale (cambiamenti rispetto alle valutazioni basali): macerazione, rossore/irritazione, eruzione cutanea/eczema, formazione di vesciche, dermatite, strappo della pelle durante la rimozione della medicazione, trauma ai bordi della ferita, degradazione del prodotto sulla pelle.
Obiettivi secondari dello studio:
- Valutazione del dolore associato alla medicazione (misurato usando una scala analogica visiva [VAS] di 100 mm).
- Stato della lesione (misurato dai cambiamenti nelle dimensioni e dalla fase di guarigione della lesione).
- Opinioni del medico/del paziente riguardo al prodotto in esame.
- Prestazioni tecniche del prodotto in studio (misurate dalla presenza di residui della medicazione in seguito alla rimozione e dalla gestione dell’essudato della ferita).
- I pazienti che presentavano più di un’ulcera erano idonei per essere inclusi nell’indagine; tuttavia, nell’indagine è stata inclusa una sola ulcera per paziente.
- Ogni paziente è stato trattato in base alla pratica clinica locale e tutte le medicazioni sono state applicate in base alle istruzioni del fabbricante.
- I pazienti hanno partecipato all’indagine fino alla completa guarigione della ferita o finché la ferita trattata non è divenuta asciutta (nel cui caso il prodotto in studio non poteva più essere applicato) oppure per 12 settimane, a seconda di quale evento si è verificato per primo.
- I pazienti sono stati valutati al basale e nuovamente alle settimane 1, 2, 4, 6, 8 e 12 post-trattamento.
- I cambi delle medicazioni sono stati effettuati in base alla pratica clinica locale (in genere 3 volte alla settimana) quando la medicazione era satura e in base allo stato e alla sede della ferita; i cambi delle medicazioni tra le visite erano consentiti.
- A ogni visita di controllo, sono state valutate le seguenti variabili:
- Dimensione e volume della ferita (usando il programma Pictzar) e profondità nel punto più profondo della ferita (usando un cotton fioc); le misurazioni sono state condotte ogni quattro settimane e una misurazione aggiuntiva è stata effettuata alla settimana 6)
- Aspetto del letto della lesione
- Condizione della cute perilesionale
- Performance tecniche
- Quantità e tipo di essudato della ferita
- Necessità di sbrigliamento
- Eventi avversi (AE) / effetti avversi del dispositivo (ADE)
- Dolore (prima della rimozione della medicazione secondaria; prima della rimozione del prodotto in esame; durante la rimozione del prodotto in esame e dopo la rimozione del prodotto in esame). Alcuni pazienti non erano eleggibili per essere inclusi nella valutazione del dolore a causa di neuropatia
- Segni clinici di infezione
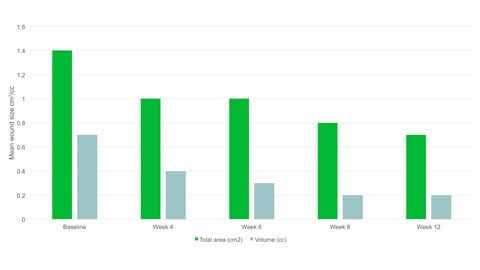
Risultati
- Nell’indagine sono stati inclusi 21 pazienti provenienti da due centri.
- Tutti i pazienti reclutati (eleggibili per il trattamento con il prodotto sperimentale) erano maschi caucasici con una DFU attiva; l'età media dei pazienti era di 59,9 anni.
Condizione della cute perilesionale
- Il numero di pazienti con una cute perilesionale sana/intatta è aumentato da 6 pazienti (28,6%) al basale a 14 pazienti (66,7%) alla visita finale.
- La percentuale di pazienti con un determinato segno di cattivo stato della cute perilesionale è diminuita o è rimasta pari a zero per la maggior parte dei segni, dal basale alla visita finale.
- Non vi sono state occorrenze di degradazione del prodotto sulla cute.
Essudato della ferita
- È stata osservata una costante riduzione del volume di essudato della ferita lungo tutto il periodo dello studio; la percentuale di pazienti senza essudato della ferita è aumentata dallo 0% al basale al 33,3% alla settimana 12.
- L’essudato della ferita era di natura prevalentemente sierosa lungo tutto il periodo dello studio.
Dolore
- I livelli di dolore erano molto bassi per tutto il periodo dello studio (alcuni pazienti non erano eleggibili per essere inclusi nella valutazione del dolore a causa di neuropatia o della guarigione completa dell’ulcera).
Stato della ferita
- Entro la settimana 12 l’area media della ferita si era dimezzata rispetto al basale, con un’area media di 0,7 cm2.
- È stata osservata una riduzione significativa dell'area lesionale, p=0,094 e del volume della ferita, p=0,0056 dal basale alla visita finale.
- È stato registrato inoltre un graduale aumento della percentuale media di tessuto di riepitelializzazione e una lieve diminuzione della percentuale media di tessuto di granulazione lungo tutto lo studio; la percentuale di tessuto non vitale è rimasta bassa durante tutto lo studio.
- Il numero di ferite guarite è aumentato da 1 (4,8%) alla settimana 1 a 5 (23,8%) alla settimana 12.
Segni clinici di infezione
È stato osservato un basso livello di segni clinici di infezione lungo tutto il periodo dello studio.
Valutazione da parte del medico/infermiere
La medicazione oggetto di valutazione è stata giudicata molto positivamente in termini di facilità di applicazione, facilità di rimozione, assenza di adesione al letto della ferita e pelle sana intatta al momento della rimozione, conformabilità, adattabilità, capacità di assorbire l’essudato, capacità di trattenere il tessuto necrotico e il sangue ed esperienza complessiva.
Valutazione da parte dei pazienti
La medicazione oggetto di valutazione è stata giudicata molto positivamente in termini di assenza di ansia durante il cambio della medicazione, facilità di movimento e stabilità della medicazione durante l’utilizzo, assenza di fitte dolorose o bruciore durante l’utilizzo e comfort.
Durante il corso dello studio non sono stati segnalati AE/ADE giudicati correlati al prodotto sperimentale.
Conclusioni
- I risultati del presente studio hanno dimostrato la capacità della medicazione in esame di ridurre al minimo il danno alla cute perilesionale e il dolore associato alla medicazione.
- Sebbene la maggior parte delle ferite non sia guarita entro il termine dello studio, sono stati notati dei miglioramenti nel tipo di tessuto e una riduzione significativa dell’area e del volume della ferita.
- Le prestazioni tecniche della medicazione sono state dimostrate dalla capacità di assorbire e trattenere l'essudato.
- La sicurezza del prodotto è stata confermata da un aumento del numero di pazienti con cute perilesionale sana/intatta e dall'assenza di eventi avversi (AE) / effetti avversi del dispositivo (ADE) identificati correlati al prodotto.
Prodotti Mölnlycke correlati

Exufiber
Medicazione in fibra gelificante con tecnologia Hydrolock® per proprietà avanzate di ritenzione dei fluidi per ferite da moderatamente ad altamente essudanti e lesioni cavitarie

Mepilex Border Flex
Medicazione "all-in-one" progettata per rimanere applicata e adattarsi in modo unico

Mepilex Border
Medicazione "all-in-one" a cinque strati per le ferite acute e croniche. Scopri l'esclusiva conformabilità della tecnologia Flex.

Mepilex Border Heel

Mepilex Border Sacrum



